--przyczyny-objawy-i-leczenie.jpg)
L'alcaptonurie (maladie protectrice) est une maladie dans laquelle le corps ne décompose pas deux des acides aminés phénylalanine et tyrosine. En conséquence, le produit de leur transformation - l'acide homogentisique - s'accumule dans le corps, conduisant, entre autres, à aux changements de peau pigmentés, à l'arthrite et aux tendinites et, dans les cas plus graves, à une insuffisance rénale ou à des lésions des valves cardiaques Quelles sont les causes et les symptômes de l'alcaptonurie? Quel est le traitement?
L'alcaptonurie (protection), communément appelée maladie de l'urine noire, est une maladie métabolique génétiquement déterminée au cours de laquelle le métabolisme de deux acides aminés est perturbé - la phénylalanine et la tyrosine. La conversion de ces composés n'est pas complète mais est arrêtée au stade de l'acide homogentisique. En conséquence, ce composé s'accumule dans l'organisme sous forme de polymères, qui au fil du temps conduisent à une décoloration des tissus (savamment appelée «protection») et à leurs dommages. L'acide homogentisique est le plus souvent déposé dans la peau, le cartilage, le fascia, les parois vasculaires, les poumons, la sclérotique, les valves cardiaques, la dure-mère et le tympan.
L'alcaptonurie est diagnostiquée chez une personne sur 100 à 250 000. dans le monde, mais dans certaines régions, il est plus fréquent, comme dans le nord-ouest de la Slovaquie (où la prévalence est estimée à environ 1 personne sur 20 000) et en République dominicaine, dans certaines régions de la Jordanie et du sud de l'Inde.
En outre, la maladie est deux fois plus fréquente chez les hommes que chez les femmes et est plus fréquente chez les enfants de parents apparentés.
Écoutez l'alcaptonurie. Il s'agit de matériel du cycle BON ÉCOUTE. Podcasts avec des conseils.Pour visionner cette vidéo, veuillez activer JavaScript et envisager de passer à un navigateur Web prenant en charge la vidéo
Alcaptonurie - causes
La maladie est causée par une carence en une enzyme appelée acide homogentisique oxydase (HGO), qui est impliquée dans la conversion de la tyrosine et de la phénylalanine et est responsable de la dégradation de l'acide homogentyisique dans l'organisme. Cette carence est causée par une mutation du gène HDG, qui contient des instructions pour la production de ces enzyme.
La maladie est héritée de manière autosomique récessive, c'est-à-dire que pour qu'un enfant tombe malade, il doit recevoir une copie du gène défectueux de chaque parent.
Alcaptonurie - symptômes
Pendant l'enfance, les seuls symptômes de la maladie sont un changement de la couleur de l'urine de la paille au noir ou une teinture dans cette couleur des couches et des sous-vêtements.
La forme à part entière de la maladie n'apparaît qu'à l'âge de 30 à 40 ans. Ensuite, les éléments suivants apparaissent:
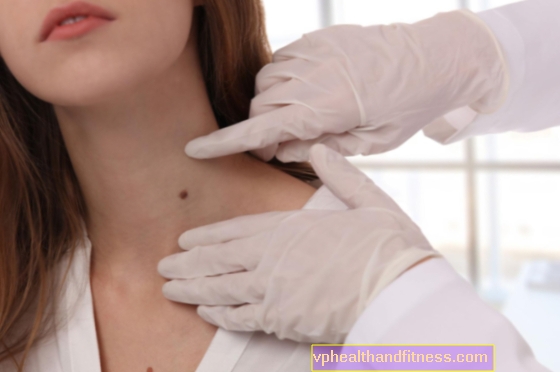
- changements sur la peau - taches jaunes, jaune-brun, marron, brun foncé ou gris-bleu. Ils sont de tailles différentes et apparaissent sur différentes zones du corps:
- oreillettes - des taches marron foncé ou gris-bleu y apparaissent très souvent. Ensuite, le symptôme qui l'accompagne est du cérumen foncé ou même noir
- visage - dans la zone de l'arcade zygomatique, la peau devient brune ou foncée
- paupières - des taches noires apparaissent
- membres - des taches apparaissent sur les doigts, sur les plaques de l'ongle, sur la plante des pieds
- poitrine - une décoloration gris-bleu de la peau est perceptible
- aisselles, anus et organes génitaux - la couleur brun-olive apparaît
- sur les tendons, le plus souvent autour des poignets
Un symptôme caractéristique de l'alcaptonurie est le changement de couleur de l'urine en noir au contact de l'air
- modifications du système ostéo-articulaire
- la dite arthropathie protectrice, c'est-à-dire modifications dégénératives des articulations, affectant généralement les grosses articulations (épaule, hanche et genou), qui se manifestent, entre autres, par douleurs articulaires, raideur matinale, mobilité limitée des articulations et leur déformation
- sensation de raideur et de douleur dans la région sacro-lombaire de la colonne vertébrale, qui résulte de la destruction des disques et de la calcification des vertèbres, en particulier dans la colonne lombaire
- approfondissement de la cyphose thoracique, avec réduction simultanée de la lordose lombaire
- Troubles auditifs - sont le résultat de l'accumulation d'acide homogentisique dans le tympan, ce qui entraîne une perte auditive ou une surdité
- taches noires sur le globe oculaire - là où la sclérotique rencontre la cornée
- néphrolithiase et altération de la fonction rénale
- calcifications des artères coronaires et lésions des valves aortiques
- chez l'homme, calcifications de la prostate
Alkaptonurie - diagnostic
Si une alcaptonurie est suspectée, une chromatographie en phase gazeuse est effectuée - une étude qui vous permet de détecter l'acide homogentisique dans l'urine.
De plus, des radiographies de la colonne vertébrale (montrant une dégénérescence discale et des calcifications de la colonne lombaire) et des articulations sont nécessaires.
Alkaptonurie - traitement
Le patient peut recevoir de fortes doses d'acide ascorbique (vitamine C), ce qui peut limiter le dépôt du pigment protecteur dans les tissus. Cependant, la découverte la plus récente dans le traitement de l'alcaptonurie est la préparation NTBC, un inhibiteur de la dioxygénase d'acide 4-hydroxyphénylpyruvique qui réduit la production d'acide homogentisique.
Jusqu'à récemment, les médecins recommandaient un régime pauvre en protéines pour inhiber la production d'acide homogentisique. Cependant, il s'est avéré que son utilisation à long terme n'a pas apporté les résultats escomptés.
De plus, un traitement symptomatique est utilisé, c'est-à-dire des anti-inflammatoires non stéroïdiens, une kinésithérapie, une thérapie physique, des massages.
Dans les modifications dégénératives très avancées des articulations, il peut être nécessaire d'insérer des implants (arthroplastie). L'intervention d'un chirurgien peut également être nécessaire en cas de lésion de la valve mitrale.
Lisez aussi: Maladie du sirop d'érable (MSUD) - Causes, symptômes et traitement Le syndrome de Smith-Lemli-Opitz est une maladie métabolique rare. Est un traitement pour SL ... Phénylcétonurie - maladie métabolique congénitale-objawy-jak-rozpozna-otpienie-po-udarze.jpg)