L'homocystinurie est une maladie métabolique rare et génétiquement déterminée dans laquelle le métabolisme de l'acide aminé méthionine est anormal. Quelles sont les causes et les symptômes de l'homocystinurie? Est-il possible de le traiter?
L'homocystinurie est une maladie génétique héréditaire de manière autosomique récessive et est le plus souvent causée par une mutation du gène CBS, et moins fréquemment par des mutations des gènes MTHFR, MTR, MTRR et MMADHC.
Cette mutation se produit à une fréquence de 1: 160 000 naissances. La maladie affecte les hommes comme les femmes. Le plus souvent, il se produit en Irlande, en Allemagne, en Norvège, au Qatar.
L'homocystinurie est la présence d'une carence en cystathionine bêta-synthase (mutation dans le gène CBS, environ 160 mutations différentes ont été enregistrées) dans le foie ou une perturbation de la conversion de l'homocystéine en méthionine (mutations dans les gènes), qui se traduit par une augmentation du taux d'homocystéine dans le sang et l'urine et une augmentation du taux de méthion . La cystathionine est une enzyme impliquée dans la réaction qui convertit l'homocystéine en cystéine, avec la participation de la pyridoxine (vitamine B6).
L'accumulation d'homocystéine dans le sang est toxique pour l'homme. L'épithélium est endommagé, provoquant des anomalies dans le tissu conjonctif, le tissu osseux, le système nerveux central et les tissus oculaires.
De plus, il existe des troubles de la coagulation, des caillots sanguins dans les vaisseaux apparaissent, entraînant souvent une embolie pulmonaire, des crises cardiaques, des accidents vasculaires cérébraux, etc.
Les personnes atteintes d'homocystinurie sont sujettes à une carence en acide folique et en vitamine B 12.
Homocystinurie: symptômes
Les enfants atteints d'homocystinurie, malgré leur phénotype normal à la naissance, présentent des caractéristiques de retard de développement: ils prennent du poids, présentent un retard de croissance par rapport à leurs pairs au fur et à mesure que la maladie progresse. La maladie est variable, avec une personne bénigne et l'autre grave.
Des symptômes oculaires peuvent survenir:
- myopie
- subluxation du cristallin
- cataracte
- atrophie du nerf optique
- glaucome
- décollement de la rétine
- tremblement des iris
Les symptômes squelettiques se manifestent par des défauts posturaux et des anomalies de la structure de la poitrine (thorax convexe en forme d'entonnoir), la présence d'ostéoporose, ce qui augmente le risque de fractures pathologiques.
Membres allongés, doigts minces en forme d'araignée, pied creux, palais haut et "gothique" se développant, raideur articulaire avec tendance à se contracter. Les patients sont généralement minces et minces.
Un handicap mental et même une maladie mentale sont souvent diagnostiqués. Le niveau d'intelligence et la capacité d'apprentissage varient individuellement en fonction de la gravité de la maladie.
Des troubles de la personnalité et de l'humeur ainsi que de l'épilepsie peuvent survenir.
Certains patients développent des hernies, en particulier inguinales et ombilicales, une stéatose hépatique, de faibles taux de facteurs de coagulation, une odeur d'urine désagréable, des troubles endocriniens, une peau claire et fine sujette à une décoloration, des changements du visage sous forme d'éruption cutanée ou de rougeurs soudaines, une cyanose , anémie, pancréatite.
La cause du décès est généralement des complications thromboemboliques résultant des changements décrits ci-dessus dans le système de coagulation (généralement dans la 3e décennie de la vie)
On peut distinguer deux groupes parmi les patients atteints d'homocystinurie:
- les patients qui répondent au traitement à la pyridoxine - avec un cours plus doux; la maladie est très probablement due à une trace d'activité de l'enzyme CbS
- ne répond pas au traitement à la pyridoxine - évolution plus sévère
Homocystinurie: diagnostic
Le diagnostic d'homocystinurie comprend:
- une histoire détaillée (symptômes de l'enfant, antécédents familiaux)
- examen physique de l'enfant (caractéristiques de l'homocystinurie)
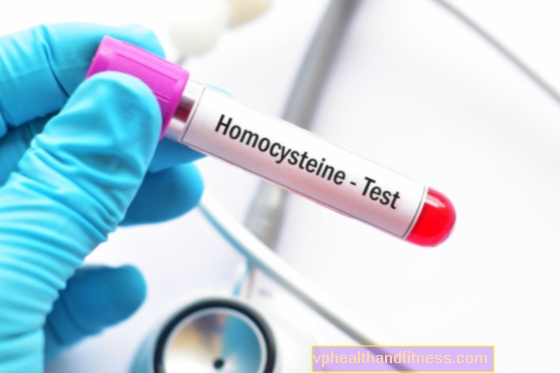
- des tests sanguins qui analysent les niveaux d'acides aminés dans le sang et l'urine, y compris l'homocystéine totale et la méthionine; test de l'activité de la cystathionine synthétase dans des cellules et tissus sélectionnés et dépistage des mutations de CBS. Les niveaux de cystéine sont généralement abaissés
En Pologne, selon le programme actuel de dépistage néonatal, le dépistage de l'homocystinurie est effectué.
- un conseil génétique doit être proposé à la famille d'une personne atteinte d'homocystinurie
- un diagnostic différentiel doit être effectué, qui doit inclure d'autres causes des malformations susmentionnées. La luxation du cristallin se produit également dans d'autres maladies, telles que le syndrome de Marfan, le syndrome de Weill-Marchesani, l'hyperlysinémie et la sulfocystéinurie. Le syndrome de Marfan doit toujours être pris en compte dans le diagnostic différentiel de l'homocystinurie car il est le plus phénotypiquement similaire
Homocystinurie: traitement
Après le diagnostic de la maladie chez le nouveau-né, un traitement précoce doit être mis en œuvre, ce qui permettra de maintenir la capacité intellectuelle et de prévenir les troubles du développement chez l'enfant. À un âge plus avancé, le traitement consiste à prévenir la thromboembolie et les complications du côté des organes.
Dans le groupe de patients qui répondent bien au traitement par la vitamine B6, des doses thérapeutiques sont administrées avec la vitamine B12 et l'acide folique.
Le deuxième groupe, insensible au traitement à la pyridoxine, suit un régime avec une teneur réduite en protéines animales et en méthionine, tout en complétant avec de la cystéine. Ce schéma thérapeutique est complété par des doses thérapeutiques de pyridoxine avec de la vitamine B12 et de l'acide folique. De plus, la bétaïne anhydre (Cystadane) est mise en œuvre, ce qui peut réduire les niveaux d'homocystéine.
Le traitement vise à corriger les anomalies biochimiques, en particulier à maintenir le taux d'homocystéine plasmatique - en dessous de 11 μmol / L, de préférence en dessous de 5 μmol / L. Cela est possible si le traitement est débuté tôt, ce qui est obtenu grâce au dépistage néonatal.
Les défauts résultant de l'évolution de l'homocystinurie doivent être traités de manière appropriée dans les spécialités appropriées.
Dans le cas d'un traitement chirurgical, le risque beaucoup plus élevé de complications thromboemboliques après anesthésie et chirurgie chez les patients atteints d'homocystinurie doit être pris en compte.
Le risque thromboembolique est élevé si le taux d'homocystéine plasmatique dépasse 50 μmol / L - l'anesthésie est alors contre-indiquée.
Avant l'intervention chirurgicale prévue, le taux d'homocystéine et de facteur VII de coagulation dans le plasma doit être vérifié et un régime pauvre en protéines et en méthionine doit être mis en place en raison de complications thromboemboliques. Un traitement anticoagulant doit être envisagé après la chirurgie.
Le risque de thrombose augmentant chez les femmes enceintes souffrant d'homocystinurie, en particulier en période post-partum, une anticoagulation prophylactique est recommandée au troisième trimestre et en période post-partum.
Habituellement, l'héparine de bas poids moléculaire est administrée par voie intraveineuse au cours des deux dernières semaines de grossesse et pendant les six premières semaines après la naissance. Il faut également envisager d’administrer de faibles doses d’aspirine pendant la grossesse.
L'homocystinurie maternelle n'affecte pas le développement de l'enfant et ne nécessite pas de surveillance plus étroite des taux plasmatiques d'homocystéine pendant la grossesse













-czyli-leczenie-kolorami.jpg)

---zasady-i-efekty-diety-orkiszowej.jpg)