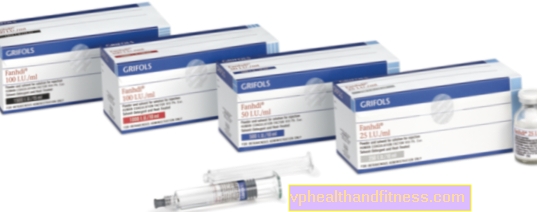
1 flacon contient 250 UI, 500 UI ou 1000 UI facteur VIII de coagulation sanguine humaine (FVIII) et 300 UI, 600 UI respectivement ou 1200 UI le facteur humain von Willebrand. L'activité spécifique est d'au moins 2,5 UI. jusqu'à 10 UI FVIII: C / mg de protéines en fonction de la taille de l'emballage (250 UI, 500 UI et 1000 UI). Produit à partir du plasma de donneurs humains.
| Nom | Contenu de l'emballage | La substance active | Prix 100% | Dernière modification |
| FANHDI | 1 flacon + seringue de 1 ampère, poudre et reconstitution pour la préparation Solution pour le choc et / ou inf. | Facteur VIII, Facteur von Willebrand | 2019-04-05 |
action
La combinaison de facteurs de coagulation - le facteur VIII et le facteur von Willebrand sont des composants physiologiques du plasma humain qui agissent comme des composants endogènes. L'administration du facteur von Willebrand permet de corriger les troubles hémostatiques survenant chez les patients présentant un déficit en facteur von Willebrand à deux niveaux: le facteur von Willebrand restabilise l'adhérence des plaquettes à l'endothélium vasculaire au site de lésion vasculaire (il se fixe à la fois à l'endothélium et à la membrane des plaquettes sanguines), ce qui entraîne la l'hémostase, qui se manifeste par une réduction du temps de saignement (l'effet apparaît immédiatement et dépend largement du niveau de polymérisation des protéines); Le facteur von Willebrand provoque une augmentation retardée du déficit en facteur VIII concomitant, une injection intraveineuse de facteur von Willebrand se lie au facteur VIII endogène (qui est synthétisé chez le patient), stabilisant ce facteur empêche sa dégradation rapide. Par conséquent, l'administration de facteur von Willebrand pur (une préparation à faible facteur VIII de facteur von Willebrand) rétablit les taux de facteur VIII à la normale en tant qu'effet secondaire après la première perfusion. Lorsqu'il est administré par voie intraveineuse à des patients hémophiles, le facteur VIII se lie au facteur von Willebrand dans la circulation sanguine du patient. Le facteur VIII actif (VIlla) agit comme un cofacteur du facteur IX actif (IXa) pour accélérer la conversion du facteur X en facteur X actif (Xa). Le facteur X actif convertit la prothrombine en thrombine. La thrombine transforme alors le fibrinogène en fibrine, ce qui permet à un caillot de se former. Chez les patients atteints d'hémophilie A, après une injection, environ deux tiers à trois quarts du facteur VIII restent dans la circulation. Le niveau d'activité du facteur VIII atteint dans le plasma doit être compris entre 80% et 120% de l'activité prévue du facteur VIII. L'activité du facteur VIII dans le plasma diminue avec une distribution diphasique exponentielle. Dans la phase initiale, la distribution entre les compartiments intravasculaire et les autres compartiments (liquides organiques) se produit avec une demi-vie d'élimination plasmatique de 3 à 6 heures. Dans la phase suivante, plus lente, qui est susceptible de refléter la consommation de facteur VIII, la demi-vie est de 8 à 18 heures, avec une moyenne de 15 heures. h.
Dosage
Par voie intraveineuse. Le traitement doit être instauré et supervisé par un médecin expérimenté dans le traitement des troubles hémorragiques. Déficit en facteur VIII: la dose et la durée du traitement dépendent de la sévérité du déficit en facteur VIIII, de la localisation et de l'étendue du saignement et de l'état clinique du patient. 1 UI L'activité du facteur VIII équivaut à la quantité de facteur VIII dans 1 ml de plasma humain normal. Administration de 1 UI facteur VIII / kg augmente l'activité du facteur VIII plasmatique d'environ 1,7% à 2,5% de l'activité normale, la dose requise est donc calculée comme suit: nombre d'unités requis = poids corporel (kg) x augmentation souhaitée du facteur VIII (%) x 0,5 . La quantité à administrer et la fréquence d'administration doivent toujours dépendre de l'efficacité clinique individuelle dans un cas donné (parfois, par exemple en présence d'un titre d'inhibiteur faible, il peut être nécessaire d'utiliser une dose supérieure à celle calculée selon la formule). Lors des saignements et de la chirurgie, l'activité du facteur VIII au fil du temps ne doit pas tomber en dessous des niveaux d'activité suivants (en% de la normale ou UI / dL): saignement précoce dans les articulations, les muscles ou la bouche - le taux de facteur VIII requis est de 20 à 40% de la normale et le médicament est administré toutes les 12 à 24 heures pendant au moins 1 jour, jusqu'à ce que le saignement soit terminé (soulagement de la douleur) ou que la plaie soit guérie; saignements plus sévères dans les articulations, les muscles ou l'hématome - le taux de facteur VIII requis est de 30 à 60% de la normale et les perfusions de médicaments sont répétées toutes les 12 à 24 heures pendant 3 à 4 jours ou plus jusqu'à ce que le soulagement de la douleur et l'altération aiguë de la fonction soient résolus; saignements potentiellement mortels - le taux de facteur VIII requis est de 60 à 100% de la normale, les perfusions de médicaments sont répétées toutes les 8 à 24 heures jusqu'à ce que la menace soit résolue; chirurgie mineure (y compris l'extraction dentaire) - le taux de facteur VIII requis est de 30 à 60%, le médicament est administré toutes les 24 heures pendant au moins 1 jour, jusqu'à ce que la plaie soit guérie; chirurgie majeure - les taux de facteur VIII requis sont de 80 à 100% (avant et après la chirurgie), les perfusions sont répétées toutes les 8 à 24 heures jusqu'à ce que la plaie soit guérie, puis poursuivies pendant au moins 7 jours consécutifs, en maintenant les taux de facteur VIII à 30-60 % (UI / dL). La réponse au traitement varie individuellement et par conséquent, la surveillance des taux de facteur VIII est essentielle, en particulier en chirurgie majeure. Pour la prophylaxie à long terme des saignements dans l'hémophilie A sévère, la dose habituelle de facteur VIII est de 20 à 40 UI / kg. tous les 2-3 jours, des intervalles de dose plus courts ou des doses plus élevées peuvent être nécessaires chez les patients plus jeunes. Maladie de Von Willebrand. On suppose généralement que l'administration de 1 UI de VWF: RCo / kg de poids corporel augmente le taux de VWF: RCo dans la circulation de 2%. Le but du traitement est d'obtenir le taux de VWF: RCo> 0,6 UI / ml (60%) et de FVIII: C> 0,4 UI / ml (40%) dans le plasma. Dans la plupart des cas, pour l'hémostase, une dose de 40 à 80 UI / kg de poids corporel de facteur von Willebrand et de 20 à 40 UI / kg de poids corporel de FVIII: C est recommandée. Les patients atteints de la maladie de von Willebrand de type 3 qui peuvent nécessiter des doses plus élevées pour maintenir des niveaux adéquats de facteur peuvent nécessiter une dose initiale de facteur de von Willebrand de 80 UI / kg de poids corporel. La dose choisie doit être administrée toutes les 12 à 24 h. La posologie et la durée du traitement dépendent de l'état clinique du patient, de la localisation et de l'étendue du saignement, ainsi que du taux de FVW: RCo et de FVIII: C. Lors de l'utilisation de préparations de facteur VIII contenant du facteur von Willebrand, le médecin traitant doit tenir compte de la possibilité d'une augmentation excessive des taux de FVIII: C. Pour éviter une augmentation excessive des taux de FVIII: C, une réduction de la dose et / ou une extension de l'intervalle entre les doses ou l'utilisation de médicaments contenant le FVW et une quantité inférieure de facteur VIII doivent être envisagées après 24 à 48 heures de traitement. Seules des données limitées issues d'essais cliniques chez des enfants de moins de 6 ans sont disponibles pour l'indication ci-dessus - ne pas utiliser. Chez l'enfant, dans les indications précitées, la titration à l'efficacité clinique est également réalisée en calculant la dose en fonction du poids corporel. Le médicament est administré sous forme d'injection intraveineuse lente (la vitesse d'administration ne doit pas dépasser 10 ml / min).
Les indications
Pour prévenir et contrôler les saignements chez les patients atteints d'hémophilie A (déficit congénital en facteur VIII de coagulation humain). Prévention et contrôle des saignements (y compris des saignements chirurgicaux) chez les patients atteints de la maladie de von Willebrand (VWD) lorsque le traitement par desmopressine (DDAVP) est inefficace ou contre-indiqué. La préparation peut être utilisée dans le traitement du déficit acquis en facteur VIII de coagulation humain.
Contre-indications
Hypersensibilité à la substance active ou à l'un des excipients.
Précautions
Des réactions d'hypersensibilité de type allergique sont possibles. Les patients doivent être informés des premiers signes de réactions d'hypersensibilité, y compris éruption cutanée, urticaire étendue, oppression thoracique, respiration sifflante, hypotension, jusqu'au choc anaphylactique. Si de tels symptômes apparaissent, l'administration du médicament doit être interrompue immédiatement et un traitement approprié doit être instauré le cas échéant. Les patients traités par le facteur VIII de coagulation humain doivent être étroitement surveillés pour le développement d'inhibiteurs anti-facteur VIII par observation clinique et évaluation des tests de laboratoire. Le risque de développer des inhibiteurs est corrélé à la durée d'exposition au facteur VIII, le risque étant le plus élevé au cours des 20 premiers jours d'exposition. En de rares occasions, des inhibiteurs peuvent également se développer après les 100 premiers jours d'exposition. Après le passage d'un produit à base de facteur VIII à un autre, une formation récurrente d'inhibiteurs (à faible titre) a été rapportée chez des patients préalablement traités pendant au moins 100 jours avec des antécédents de formation d'inhibiteurs. Par conséquent, une surveillance attentive des patients pour la survenue d'inhibiteurs est recommandée après tout passage au médicament. Il existe un risque de complications thrombotiques lors de l'utilisation de médicaments à base de facteur de von Willebrand, en particulier chez les patients présentant des facteurs de risque cliniques ou biologiques connus. Pour cette raison, une surveillance des patients à risque est nécessaire pour détecter les premiers signes de thrombose. En outre, les recommandations des lignes directrices actuelles pour le traitement et la prévention de la thromboembolie veineuse doivent être suivies. Pendant la période de traitement par un médicament contenant du facteur VIII et du facteur von Willebrand, le médecin doit tenir compte du fait qu'un traitement prolongé peut entraîner une augmentation excessive des taux de FVIII: C. Chez les patients traités par des médicaments contenant du facteur VIII et du facteur von Willebrand, les taux de FVIII: C doivent être surveillés pour éviter la persistance de taux excessifs qui pourraient augmenter le risque de complications thrombotiques. Les patients atteints de la maladie de von Willebrand, en particulier de type 3, peuvent développer des anticorps neutralisants (inhibiteurs) du facteur de von Willebrand. Si les niveaux d'activité plasmatique de VWF: RCo attendus ne sont pas atteints ou si le saignement est contrôlé avec des doses appropriées, un examen doit être effectué pour vérifier la présence d'un inhibiteur du facteur von Willebrand. Chez les patients présentant des niveaux élevés d'inhibiteur, le traitement par facteur de von Willbrand peut ne pas être efficace et d'autres options thérapeutiques doivent être envisagées. Si une ligne veineuse centrale est nécessaire, des infections locales, une bactériémie et une thrombose du cathéter peuvent survenir. Une vaccination appropriée (contre les virus des hépatites A et B) doit être envisagée chez les patients recevant régulièrement du facteur VIII de coagulation dérivé du plasma.
Activité indésirable
Hypersensibilité ou réactions allergiques (angio-œdème, sensations de brûlure et de picotement au site d'injection, frissons, bouffées de chaleur paroxystiques, urticaire généralisée, maux de tête, éruption cutanée, baisse de la pression artérielle, somnolence, nausées, agitation, tachycardie, oppression thoracique, sensation de picotements, vomissements, respiration sifflante), entraînant dans certains cas une anaphylaxie sévère avec choc. Des augmentations de température ont rarement été observées. Les patients atteints d'hémophilie A peuvent développer des anticorps neutralisants (inhibiteurs) du facteur VIII. Lorsque de tels inhibiteurs se développent, une réponse clinique insuffisante au traitement est observée. Dans de très rares cas, les patients atteints de la maladie de von Willebrand, en particulier de la maladie de type 3, peuvent développer des anticorps neutralisants (inhibiteurs) du facteur von Willebrand. Si de tels inhibiteurs apparaissent, une réponse clinique insuffisante au traitement est observée. Ces anticorps peuvent être étroitement associés à une réaction anaphylactique. Pour cette raison, les patients qui présentent des réactions anaphylactiques doivent être testés pour les inhibiteurs. Dans de tels cas, il est recommandé de contacter un centre spécialisé pour le traitement des troubles hémostatiques. Il existe un risque de complications thrombotiques lorsque le médicament est utilisé chez des patients atteints de la maladie de von Willebrand présentant des facteurs de risque cliniques ou biologiques connus.
Grossesse et allaitement
La préparation ne doit être utilisée pendant la grossesse et l'allaitement qu'en cas de nécessité absolue (aucune expérience de l'utilisation du médicament en raison de la rareté de l'hémophilie A chez la femme).
Les interactions
Il n'y a pas d'interactions connues du syndrome FVIII / VWF avec d'autres préparations.
La préparation contient la substance: Facteur VIII, Facteur von Willebrand
Médicament remboursé: NON















-zagroeniem-dla-ycia-kobiety.jpg)